Allometry in the T Model
Run this notebook
Read the guide on setting up your computer to run Jupyter notebooks
Download
this notebookas a Jupyter notebook.
import warnings
from matplotlib import pyplot as plt
import numpy as np
import pandas as pd
from pyrealm.core.experimental import ExperimentalFeatureWarning
from pyrealm.demography.flora import Flora
from pyrealm.demography.cohorts import create_cohorts, cohort_id_generator
from pyrealm.demography.tmodel import StemAllometry
warnings.filterwarnings(
"ignore",
category=ExperimentalFeatureWarning,
)
To generate allometric predictions under the T Model, we need to define a set of cohorts:
# Create a flora with 3 PFTs with different maximum heights
flora = Flora(name=["short", "medium", "tall"], h_max=[10, 20, 30])
# Create an id generator.
cid_generator = cohort_id_generator(mode="str")
# Create the cohorts
cohorts = create_cohorts(
flora=flora,
cid_generator=cid_generator,
pft_name=np.array(["short", "medium", "tall"]),
dbh_value=np.array([0.1, 0.1, 0.1]),
n_individuals=np.array([1, 1, 1]),
)
Stem allometry
We can visualise how the stem size, canopy size and various masses of PFTs change with
stem diameter by using the StemAllometry
class. Creating a StemAllometry instance needs an existing Flora instance and an
array of values for diameter at breast height (DBH, metres). The returned class contains
the predictions of the T Model for:
Stem height (
stem_height, m),Crown area (
crown_area, m2),Crown fraction (
crown_fraction, -),Stem mass (
stem_mass, kg),Foliage mass (
foliage_mass, kg),Sapwood mass (
sapwood_mass, kg),Crown radius scaling factor (
crown_r0, -), andHeight of maximum crown radius (
crown_z_max, m).
Note that stem_height denotes the total
tree height, as used interchangeable in Li et al. (2014), rather than just the height
of the trunk below the canopy.
The DBH input can be a scalar array or a one dimensional array providing a single value for each PFT. This then calculates a single estimate at the given size for each stem.
# Calculate the allometry for the cohorts
cohort_allometry = StemAllometry(cohorts=cohorts)
cohort_allometry
StemAllometry: Allometry predictions for 3 cohorts.
The StemAllometry() class provides the
to_dataframe() method to export the
stem data for data exploration. The StemAllometry data retains the unique cohort ids
and DBH from the original cohort data.
cohort_allometry.to_dataframe().transpose()
| 0 | 1 | 2 | |
|---|---|---|---|
| cohort_ids | C_000000 | C_000001 | C_000002 |
| dbh | 0.1 | 0.1 | 0.1 |
| stem_height | 6.865138 | 8.802033 | 9.620475 |
| crown_area | 1.814782 | 2.326795 | 2.543148 |
| crown_fraction | 0.591822 | 0.758796 | 0.829351 |
| stem_mass | 5.391867 | 6.9131 | 7.555903 |
| foliage_mass | 0.233329 | 0.299159 | 0.326976 |
| fine_root_mass | 0.555323 | 0.711999 | 0.778203 |
| sapwood_mass | 4.493533 | 6.5109 | 7.335868 |
| crown_r0 | 0.261731 | 0.296362 | 0.309834 |
| crown_z_max | 5.83731 | 7.484219 | 8.180126 |
Allometry profiles
The at_dbh argument to StemAllometry can be used to generate profiles of the
allometry predictions for PFTs at different sizes. The provided values are used to
calculate predictions instead of the cohort DBH values. The allometry attributes of
predictions are then 2 dimensional arraus arranged with each cohort as a column and each
DBH prediction as a row. This makes them convenient to plot using matplotlib.
# Column array of DBH values from 0.01 to 1.6 metres
dbh_profile = np.arange(0.01, 1.6, 0.01)
# Get the predictions at those DBH values.
allometry_profiles = StemAllometry(cohorts=cohorts, at_dbh=dbh_profile)
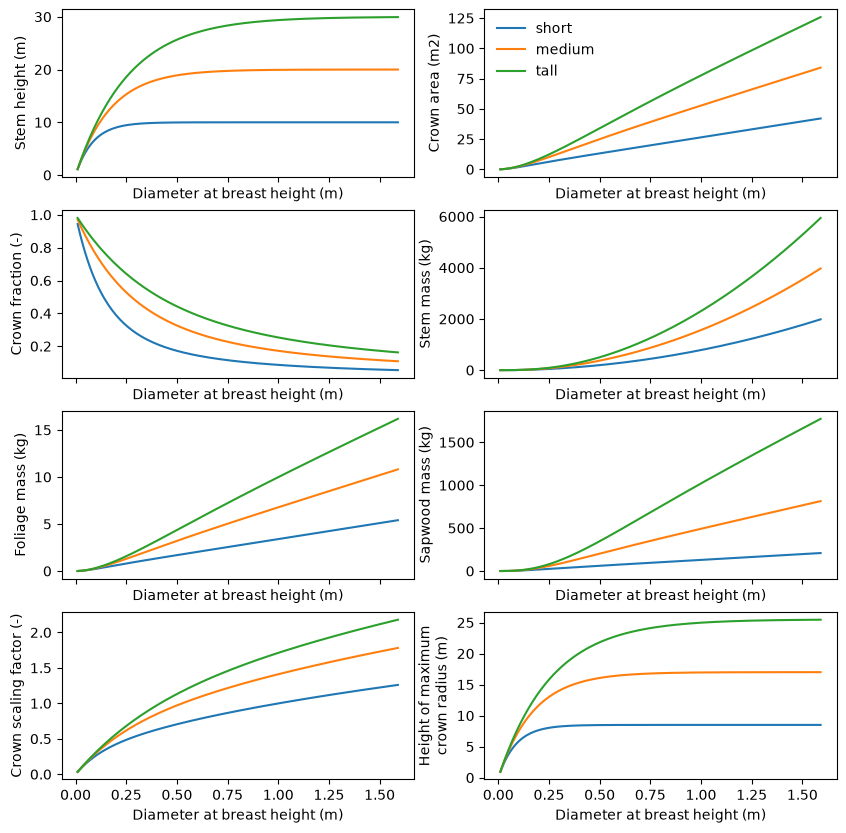
The code below shows how to use the returned allometries to generate a plot of the
scaling relationships across all of the PFTs in a Flora instance.
fig, axes = plt.subplots(ncols=2, nrows=4, sharex=True, figsize=(10, 10))
plot_details = [
("stem_height", "Stem height (m)"),
("crown_area", "Crown area (m2)"),
("crown_fraction", "Crown fraction (-)"),
("stem_mass", "Stem mass (kg)"),
("foliage_mass", "Foliage mass (kg)"),
("sapwood_mass", "Sapwood mass (kg)"),
("crown_r0", "Crown scaling factor (-)"),
("crown_z_max", "Height of maximum\ncrown radius (m)"),
]
for ax, (var, ylab) in zip(axes.flatten(), plot_details):
ax.plot(dbh_profile, getattr(allometry_profiles, var), label=flora.name)
ax.set_xlabel("Diameter at breast height (m)")
ax.set_ylabel(ylab)
if var == "crown_area":
ax.legend(frameon=False)
The to_dataframe() method can still be
used, but the values are stacked into columns identified by pairings of cohort ID and
DBH.
allometry_profiles.to_dataframe().head(6)[
["cohort_ids", "dbh", "stem_height", "crown_area", "crown_fraction"]
]
| cohort_ids | dbh | stem_height | crown_area | crown_fraction | |
|---|---|---|---|---|---|
| 0 | C_000000 | 0.01 | 1.095248 | 0.028953 | 0.944179 |
| 1 | C_000001 | 0.01 | 1.127001 | 0.029792 | 0.971553 |
| 2 | C_000002 | 0.01 | 1.137860 | 0.030079 | 0.980913 |
| 3 | C_000000 | 0.02 | 2.070539 | 0.109468 | 0.892474 |
| 4 | C_000001 | 0.02 | 2.190496 | 0.115810 | 0.944179 |
| 5 | C_000002 | 0.02 | 2.232562 | 0.118034 | 0.962311 |